Keumuman
Syarat retinitis pigmentosa (RP) mengidentifikasi sekelompok penyakit genetik yang ditandai dengan degenerasi retina progresif.

Retinitis pigmentosa adalah distrofi retina yang ditandai dengan hilangnya fotoreseptor secara bertahap dan disfungsi epitel pigmen, yang berarti retina secara progresif mengurangi kemampuannya untuk mengirimkan informasi visual ke otak melalui saraf optik.
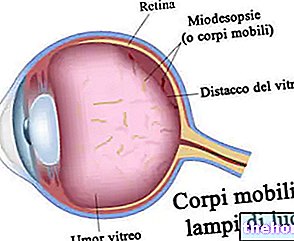
Proses patologis dimulai dengan perubahan epitel pigmen retina. Seiring berkembangnya retinitis pigmentosa, terjadi penipisan pembuluh darah yang mensuplai retina, yang mengalami atrofi. Pada pemeriksaan fundus, endapan khas dapat dideteksi secara visual. pigmen retina ( maka nama dari penyakit). Perubahan dan kerusakan atrofi juga dapat melibatkan saraf optik dan, secara bertahap, sel-sel fotosensitif retina mati.
Pasien yang terkena retinitis pigmentosa awalnya mengalami masalah penglihatan terutama di lingkungan yang kurang penerangan dan mengeluhkan penyempitan bidang visual perifer. Penglihatan sentral terhindar sampai tahap akhir penyakit, dan hasil akhirnya dapat bervariasi secara dramatis: banyak orang dengan retinitis pigmentosa mempertahankan penglihatan terbatas sepanjang hidup mereka, sementara yang lain kehilangan penglihatan sama sekali.
Retinitis pigmentosa adalah penyakit bawaan, terutama disebabkan oleh perubahan genetik yang diturunkan dari salah satu atau kedua orang tua. Jenis cacat genetik menentukan sel retina mana yang paling terlibat dalam gangguan dan memungkinkan untuk membedakan, dari sudut pandang klinis, kondisi yang berbeda. Sampai saat ini, lebih dari 50 cacat genetik berbeda yang terlibat dalam retinitis pigmentosa telah diidentifikasi. Kelainan dapat diturunkan dari orang tua ke keturunannya melalui salah satu dari tiga pola pewarisan: resesif autosomal, dominan autosomal, atau resesif heterosomal (terkait-X atau terkait-X).
Gejala
Untuk informasi lebih lanjut: Gejala Retinitis Pigmentosa
Retinitis pigmentosa biasanya ditemukan pada remaja dan dewasa muda. Gejala sering muncul antara usia 10 dan 30, tetapi diagnosis dapat dibuat pada anak usia dini atau jauh di kemudian hari.
Gejala awal retinitis pigmentosa dapat meliputi:
- Kesulitan melihat di malam hari (rabun senja) atau dalam kondisi kurang cahaya
- Adaptasi lambat dari penglihatan dalam gelap ke terang, dan sebaliknya;
- Penyempitan bidang visual dan hilangnya penglihatan tepi;
- Kepekaan terhadap cahaya dan silau.
Beberapa gejala tergantung pada jenis fotoreseptor yang terlibat. Batang bertanggung jawab untuk penglihatan hitam dan putih, sedangkan kerucut memungkinkan Anda untuk membedakan warna.
Dalam kebanyakan kasus retinitis pigmentosa, batang yang terlibat terlebih dahulu. Namun, dalam bentuk yang berkembang pesat, kerucut juga dapat terpengaruh pada tahap awal.
Batang terkonsentrasi di bagian luar retina dan diaktifkan oleh cahaya redup, sehingga degenerasinya memengaruhi penglihatan tepi dan malam. Jika sel kerucut terlibat, ada kemungkinan untuk mengalami kehilangan persepsi warna dan penglihatan sentral.
Dominasi fotoreseptor yang terlibat ditentukan oleh cacat tertentu yang ada pada susunan genetik pasien.
Seringkali, gejala pertama retinitis pigmentosa adalah rabun senja (atau nocthalopia). Beberapa orang merasa bahwa mereka membutuhkan lebih banyak waktu untuk menyesuaikan diri dengan perbedaan cahaya saat mereka berpindah dari area yang cukup terang ke area yang lebih gelap. Bentuk khas kehilangan penglihatan menyebabkan penyempitan penglihatan tepi (penglihatan terowongan atau teleskop); pola ini disebut skotoma cincin. Kadang-kadang, fenomena ini mungkin hilang pada tahap awal, tetapi diperhatikan ketika individu sering tersandung objek atau terlibat dalam kecelakaan lalu lintas Ketika kehilangan penglihatan melibatkan area pusat retina (juga disebut distrofi makula) pasien mengalami kesulitan membaca dan pekerjaan mendetail yang membutuhkan konsentrasi pada satu objek, seperti memasukkan benang melalui lubang jarum Banyak pasien melaporkan melihat kilatan cahaya (photopsia), sering digambarkan sebagai lampu kecil, berkelap-kelip dan berkelap-kelip.
Tingkat perkembangan penyakit dan tingkat kehilangan penglihatan bervariasi dari orang ke orang. Beberapa kasus ekstrem dapat berkembang pesat dalam dua dekade, yang lain perjalanan lambat yang tidak pernah menyebabkan kebutaan total. Onset dini ditemukan pada bentuk retinitis pigmentosa yang lebih parah, sementara pasien dengan kondisi yang lebih ringan (misalnya, autosomal dominan) dapat mengembangkan penyakit pada dekade kelima atau keenam kehidupan.Pada keluarga dengan retinitis pigmentosa terkait-X, pria lebih sering terkena. daripada wanita dan lebih parah; wanita, di sisi lain, mentransmisikan karakteristik genetik (mereka membawa gen yang diubah pada kromosom X) dan menunjukkan gejala gangguan lebih jarang.
Komplikasi
Retinitis pigmentosa akan terus berkembang, meskipun lambat. Namun, kebutaan total jarang terjadi, tetapi penurunan signifikan dalam penglihatan perifer dan sentral dapat terjadi.
Pasien dengan retinitis pigmentosa sering mengalami pembengkakan retina (edema makula) atau katarak pada usia dini. Komplikasi ini dapat diobati jika mengganggu penglihatan.
Penyakit terkait
Umumnya, pasien dengan retinitis pigmentosa tidak memiliki kelainan lain dan dalam hal ini kita berbicara tentang "non-sindrom" atau retinitis pigmentosa sederhana. Namun, beberapa sindrom berbagi beberapa gejala klinis dengan penyakit mata ini; yang paling umum adalah sindrom Usher, yang mempengaruhi sekitar 10-30% dari semua pasien dengan retinitis pigmentosa dan berhubungan dengan gangguan pendengaran kongenital atau progresif. Dalam amaurosis bawaan Leber, bagaimanapun, anak-anak bisa menjadi buta, atau hampir buta, dalam enam bulan pertama kehidupan.Penyakit lain yang berhubungan dengan retinitis pigmentosa termasuk sindrom Bardet-Biedl dan penyakit Refsum.
Penyebab
Penyakit ini dapat disebabkan oleh sejumlah cacat genetik: pada kenyataannya, ada beberapa gen yang, jika terpengaruh oleh perubahan, dapat menyebabkan fenotip retinitis pigmentosa.Ini biasanya mengkodekan protein yang terlibat dalam kaskade transduksi yang memungkinkan penglihatan, faktor transkripsi sel (yang mengirim pesan yang salah ke sel retina) atau untuk elemen yang membentuk struktur fotoreseptor. Mutasi gen yang diturunkan ada dalam sel sejak saat pembuahan; kelainan umum termasuk gen RP1 (pada retinitis pigmentosa-1, autosomal dominan) , RHO (RP4, autosomal dominan) dan RDS (RP7, autosomal dominan).Penyebab retinitis pigmentosa non-herediter jarang terjadi, tetapi kemungkinan menemukan kasus terisolasi (mutasi spontan), di mana tidak ada riwayat keluarga penyakit.
.jpg)